All published articles of this journal are available on ScienceDirect.

An Updated Review of Nicotine in Gastrointestinal Diseases
Abstract
Background:
Nicotine addiction is a major risk for the population all over the world. Many gastrointestinal diseases are caused due to nicotine addiction, like peptic ulcers, inflammatory bowel diseases, and gastrointestinal cancer. Nicotine causes variations in the offensive and defensive factors, which are involved in gastrointestinal mechanisms.
Objective:
We aim to provide insights into nicotine activity in gastrointestinal disorders and cancer.
Methods:
Extensive literature was done using the keywords “Nicotine,” “Nicotine addiction,” “GI disorders,” “Peptic ulcer,” “Inflammatory bowel diseases,” and “Gastrointestinal cancer” from standard databases like PubMed, Scopus, Elsevier, and Science Direct.
Conclusion:
Thus, we may conclude that addiction to nicotine in peptic ulcers causes an increase of offensive factors and a decrease of defensive factors, and in gastrointestinal cancers, causes an increase in angiogenesis, metastasis, and proliferation and decrease in apoptosis in ulcerative colitis. It is used as treatment but worsens the condition of Crohn’s disease.
1. INTRODUCTION
One very important issue that has existed in the globe for a very long time is nicotine addiction. Nicotine fuels tobacco addiction, which is a leading cause of disability and untimely death [1]. Over 4,000 chemicals are present in the tobacco smoker's environment, making it challenging to pinpoint the specific compounds that cause smoking's wide range of consequences, both negative and positive [2]. Nicotine, which is produced from tobacco plants, is an addictive and dangerous chemical that leads to both mental and physical dependence [3]. Despite this, many people have developed an addiction to using chemicals related to nicotine. Neal L. Benowitz claims that 35,000 people die annually in the United States from ailments linked to nicotine [4]. One death out of every five is a result of smoking or nicotine addiction. Most youth and students in today's society are addicted to nicotine, and this is true when they attend university or school for higher studies. Nicotine dependence is most prevalent in people between the ages of 16 and 25 [5]. In the earlier days, nicotine and nicotine-containing products helped people cope with a variety of emotions, including tension and anxiety, which frequently led to nicotine addiction [6]. Due to this, people often experience symptoms of COPD, neurological disorders, gastrointestinal problems, and cardiac disease. In many parts of the world, it is linked to severe morbidity and mortality. Therefore, nicotine addiction is a serious public health issue [7]. This review focuses, as shown in the graphical abstract, primarily on the gastrointestinal system's diseases associated with nicotine addiction as well as the mechanisms of action, pharmacokinetics, and metabolism of the drug. Among these, gastrointestinal cancer, Chron disease, inflammatory bowel diseases, gastrointestinal reflux disorders, and peptic ulcers are the most common [8].
2. EFFECT OF NICOTINE ON THE GASTROINTESTI-NAL SYSTEM
Acknowledging how nicotine affects ulcer formation and repair in the GI mucosa through both physiological and pathological processes has advanced significantly over the past 20 years [9]. In order to achieve this goal, nicotine promoted apoptosis while decreasing cell growth, blood vessel development, and mucus creation in the GI tract. Numerous studies indicate that tobacco is hazardous to the gastrointestinal tract in contrast to the impact it has on the heart and lungs [10].
2.1. Peptic Ulcer
The eroding of the mucosal lining in the gastrointestinal tract canal by stomach acid is what leads to peptic ulcers. The stomach and duodenum are the two main sites where peptic ulcers develop [11]. Prior to the first half of the 20th century, stress, dietary variables, and a very acidic environment were thought to be the primary causes of peptic ulcers. But by the middle of the 20th century, the widespread use of NSAIDs and the Helicobacter pylori infection had drastically altered our understanding of the causes of peptic ulcers [12]. Peptic ulcers can be caused by Helicobacter pylori infection, excessive use of NSAID as well as alcohol consumption, and minor physical damage during ingestion. One of the main factors contributing to peptic ulcers is cigarette smoking [13].
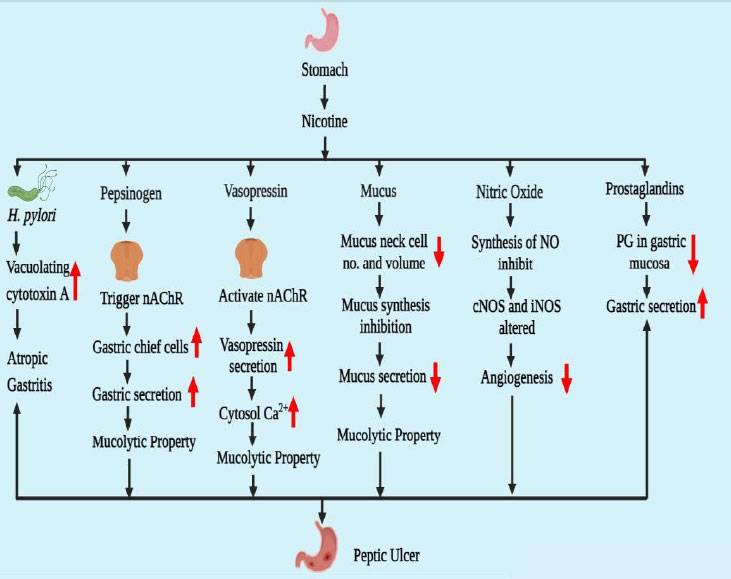
The primary component of cigarettes is nicotine, which significantly contributes to the development of peptic ulcers by disrupting the balance between offensive and protective elements in the gastrointestinal tract [14]. Gastric acid, Helicobacter pylori, pepsinogen, and vasopressin are some of the offensive factors, whereas mucosal blood flow, nitric oxide, prostaglandins, and mucus secretion are some of the protective factors that, in a balanced environment, may assist in healing the peptic ulcer [9]. The pathophysiology of peptic ulcers caused by smoking is still not entirely understood. Certain circumstances cause a rise or reduction in stomach acid output [15].
2.1.1. Role of Offensive Factors in Peptic Ulcer
2.1.1.1. Helicobacter Pylori
Despite the absence of peptic ulcer disease, nicotine can raise the chance of H. pylori infection. Smokers have a higher risk of developing H. pylori infection, which raises their risk of developing a peptic ulcer [16]. In 1967, the Germans were the first to examine the relationship between nicotine and gastrointestinal disease after observing an increase in peptic ulcers in rats due to nicotine's increased gastric juice output. In order to initially colonize and then persist in the stomach of the host, H. pylori produces a number of protein-rich substances [17]. One of the main proteins secreted by H. pylori is the cytotoxic protein known as vacuolating cytotoxin A. The crucial element of H. pylori that is triggered by nicotine is vacuolating cytotoxin A. According to this process, peptic ulcers are mostly caused by a combination of nicotine and H. pylori, which also causes a delay in peptic ulcer recovery [18].
2.1.1.2. Pepsinogen
In the presence of gastric acid, pepsinogen-a biologically inactive proenzyme converts to pepsin. Pepsinogen-I and Pepsinogen-II are the two variants of pepsinogen [19]. The muscarinic receptor (M1, M3) uses neuronal nicotinic acetylcholine receptors (nAChRs) to mediate pepsinogen secretion. Nicotine has the ability to trigger the nAChRs in gastric chief cells, and nicotine also increases the number of gastric chief cells [20]. These were the principal triggers for pepsinogen's release. Pepsinogen is a highly provocative agent in the etiology of peptic ulcers and has mucolytic properties.
2.1.1.3. Vasopressin
Vasoconstriction may result from endogenous vasopressin, which is secreted from the vascular endothelium and the posterior pituitary. Due to the Brattleboro homozygous vasopressin-deficient rat's reduced sensitivity to several ulcerogenic stimuli compared to the rat with normal vasopressin secretion, endogenous vasopressin has been shown to operate as an aggressive factor towards the gastroduodenal mucosa [21]. Nicotine and cigarette smoking promote the release of vasopressin by activating central nicotinic cholinergic projections to the hypothalamus. The release of vasopressin from neurons occurs when nicotine raises the cytosolic Ca2+ level. At least in part, cAMP-dependent protein kinase A is responsible for this action of nicotine. The release of vasopressin from the neurons is facilitated by the elevated cytosolic Ca2+ [22].
Various factors involved in causing peptic ulcer due to addiction to nicotine are represented in Fig. (1).
2.1.2. Role of Protective Factors in Peptic Ulcer
2.1.2.1. Mucus Synthesis
These epithelial cells, which make up the gastric mucosa, are in charge of sequestering mucus. The stomach mucosa was protected by mucus from both endogenous and exogenous influences. A gelatinous, water-insoluble material called gastric mucus is present in the stomach lumen [23]. Smokers have a lower level of mucus in their stomachs, and cigarette smoke or its extract markedly decreases mucus synthesis in vivo and ex vivo, both of which could be completely reversed by the application of epidermal growth factor (EGF) [8]. The protein backbone has O-linked oligo-saccharides, and n-glycan chains are attached in heavy numbers, which are high molecular weight glycoproteins and are known as mucins. Twenty-one mucin genes (MUC) have been identified in the human genome [24]. According to these genes, mucin is split into two sorts of groups: membrane-bound mucins and secreted mucins. The three most prevalent mucins found in the stomach are MUC 1 (membrane-bound), MUC5AC, and MUC6 (secreted mucins). Mucus production and secretion serve as protective mechanisms, but nicotine alters them [25]. Nicotine has two distinct mucus secretion behaviors. When supplied in small doses, nicotine stimulates mucus output, but when administered in larger doses, nicotine reduces the volume and quantity of mucus neck cells, which inhibits mucus secretion as well. Nicotine may not only negatively impact mucosal EGF affirmation as well as the production of NO and VEGF throughout the recovery of peptic ulcer [26].
2.1.2.2. Nitric Oxide
Access to cigarette smoke slowed ulcer healing and reduced angiogenesis and gastric blood flow near the ulcer border. Foundational nitric oxide synthase (cNOS) activity was significantly reduced in conjunction with these alterations [27]. A series of isozymes, including neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS), are responsible for the production of NO. Additionally, it effectively induces the release of gastrointestinal mucus by stimulating the soluble guanylate cyclase. Furthermore, investigations conducted both in vivo and in vitro have supported the roles played by NO in inducing angiogenesis [28]. Nicotine could harm endothelial cells or decrease the function of cNOS at the ulcer site (mostly granulation tissues) and the surrounding area, which would impede NO generation. Interestingly, treatment of L-arginine [29], from which NOS produced NO, totally reversed the negative effects of cigarette smoke on ulcer healing, gastric blood flow, and angiogenesis in the mucosa at the ulcer boundary but only fully recovered angiogenesis in granulation tissues [30].
2.1.2.3. Prostaglandin E2 (PGE 2)
Prostaglandins enhance mucosal blood flow, mucosal secretion, and bicarbonate synthesis while decreasing stomach acid output. Prostaglandin E2's health benefits are lessened by nicotine because it lowers the amount of this substance in the gastric mucosa. Decreased glutathione, a free radical scavenger with a high concentration in the gastric mucosa, helps to lessen cellular damage brought on by oxidative stress. It has been demonstrated that nicotine makes ulceration from stress-induced reduction in stomach glutathione levels worse [31].
2.2. Inflammatory Bowel Disease
Since the beginning of civilization, Inflammatory Bowel Disease (IBD) has occasionally been noted. In modern industrialized countries, it has become a serious and major concern. IBD was generally uncommon in underdeveloped countries, whereas its prevalence in the West countries progressively increased throughout the 20th century [32]. The rise of IBD has been reported over the last few decades in newly industrialized nations in South America, Asia, and the Middle East. The frequency of the increase in the prevalence of IBD is noticeably faster in these recently industrialized nations, but the prevalence of IBD there is still significantly lesser than in the West [33]. On the epidemiological data of IBD, numerous researches have been done. IBD has a very variable different geographical occurrence [34].
The digestive system is regularly challenged by a wide range of antigens that are included in meals and microorganisms. Gastrointestinal homeostasis is preserved and controlled by reducing exaggerated immune reactions to exogenous antigens when there is no gastrointestinal inflammation [35]. A multifactorial condition known as IBD is recently brought on by persistent, severe gastrointestinal inflammation. Alteration in the microbiota is the major reason for IBD progression. Alteration in the microbiota depends upon two factors: first, shift in taxonomic makeup, such as lower-alpha diversification, lower expression of firmicutes and bacteroides, higher frequency of adhering invaders, decreasing quantities of Faecalibacterium prausnitzii, higher incidence of Gammaproteobacteri, and second is a microbial function such as diminished short-chain fatty acid generation and amino acid synthesizing, enhanced oxidative stress, elevated sulfate transportation, and steadily increasing auxotrophy [36]. These two variables are affected by a number of exogenous variables, including smoking, pregnancy, lifestyle, genetics, medicines, and many others [37].
The digestive tract is affected by an array of inflammatory illnesses known as inflammatory bowel disease (IBD), which are regulated by the immune system. Crohn's disease (CD) and ulcerative colitis (UC) are the two primary different types of IBD (CD) [38]. In contrast to UC, which only affects the colon mucus-secreting cells and nearly always includes the rectum, every region of the GI tract is affected by CD, including the mouth and the antrum. The two of these subtypes are classified as M1- and M2-mediated actions. The main ingredient in tobacco smoking, nicotine, has inconsistent impacts on IBD [39].
2.2.1. Ulcerative Colitis
The mucosal layer of the rectum and the colon's proximal portion constantly become involved in the development of ulcerative colitis, a chronic inflammatory illness. There are three forms of UC depending on where the inflammation resides: Protitis, a left-side colitis extending to the flexura sinistra, and unusual cases of pancolitis with extra backwash ileitis [40]. There has been a global UC problem. UC is a chronic, incurable illness that impairs both physical and psychological well-being. Although the primary origin of this condition is unknown, recent research indicates that in 8 to 14% of individuals, genealogy is a key risk factor [41]. The two age groups of persons harmed by the UC condition: It takes between 15 and 30 years for the primary onset to manifest and between 50 and 70 years for the second onset. UC exhibits a bimodal pattern of the incident as an outcome. Most cases of UC are reported from western and northern countries. UC is brought on by our way of life, specifically the Western way of living [42]. Genetic and environmental factors, along with a number of additional factors like infection, autoimmunity, ischemia, toxin exposure, and immunodeficiency, all contribute to the development of UC. Patients with UC frequently have bloody stools, watery diarrhea, tenesmus, stomach pain, and urgency. The incidence of UC is reduced by 69% after appendectomy, while the chance of Chron's disease is increased [43]. This makes appendectomy a significant factor in cases of UC. Leukocyte mobilization, mucosal barriers, immunological reactivity, and colonic microbiota are all changed, which contributes to ulcerative colitis [44]. The mucin coating covering the epithelium of the stomach serves as the body's primary layer of defense and has antibacterial capabilities; however, in UC, mucin release and production are affected as a result of exposure to certain foods, microorganisms, and environmental factors [45]. When the first line of defense is damaged, the gut immune response is stimulated because lumen antigen uptake is elevated. Due to the stimulation of the inflammation response in UC, cells that contain antigens, such as macrophages and dendritic cells, activate and differentiate CD4+ T lymphocytes. Two types of T helper cells, Th-1 and Th-2, are formed from T lymphocytes [46]. Th-1 cells release interferon gamma (INFγ) and interleukin, which are IL-2, IL-12, and IL-18, while Th-2 cells release IL-4, IL-5, IL-6, IL-10, and IL-13. When the Th-1 reactions in macrophages are triggered, TNFα is generated by the macrophages. The Th-2 response is characterized by natural killer cells (NKT) that are present in CD4 cells and express intracellular IL-13 [47]. Flowing leukocytes and IL-13 both promote tight junction inflammation and epithelial cell death, which both contribute to inflammation. Anti-inflammatory drugs (5-aminosalicylates, corticosteroids), immunosuppressants (azathioprine, mercaptopurines, cyclosporins), tumor necrosis factor inhibitors (Infliximab, adalimumab), anti-diarrheal, pain relievers, antispasmodics, and other medications are several of the treatment options for ulcerative colitis [48]. Various factors involved in causing ulcerative colitis due to addiction to nicotine are represented in Fig. (2).
Several studies revealed that nicotine is also used as a treatment therapy for UC owing to the fact nicotine users have a lower incidence of UC. Through different mechanisms, nicotine alters intestinal permeability, endogenous glucocorticoids, rectal blood flow, eicosanoids, and mucus formation. These factors significantly lower the incidence of UC [49]. Nicotine exhibits the ability to inhibit both innate and adaptive immunity. The intestinal lavage IgA concentration is lowered by nicotine. It has been demonstrated in numerous in vivo and in vitro investigations that nicotine reduces the levels of IL-1B, IL-2, IL-8, IL-10, and TNF-α in colorectal mucosa and blood mononuclear cells [50]. Dendritic cells and macrophages both have nAChR, which mediates inflammation. TNF-α is decreased as a result of nicotine's stimulation of the nAChRα7 on macrophases. Even though the accurate mechanism is mysterious, the presence of nAChRα3 in colon epithelial cells enhanced cell function [51]. Dendritic cells, which are affected by nicotine, also affect the differentiation of T-helper cells. Eicosanoids such as 15-hydroxy eicosatetraenoic acid (15 HETE) and 6-keto Prostaglandin PGF1, PGF2, and PGF2 are decreased by nicotine on the rectal mucosa but not the caecal mucosa. NO is incredibly significant in the UC case. Nicotine increases the levels of NO in the
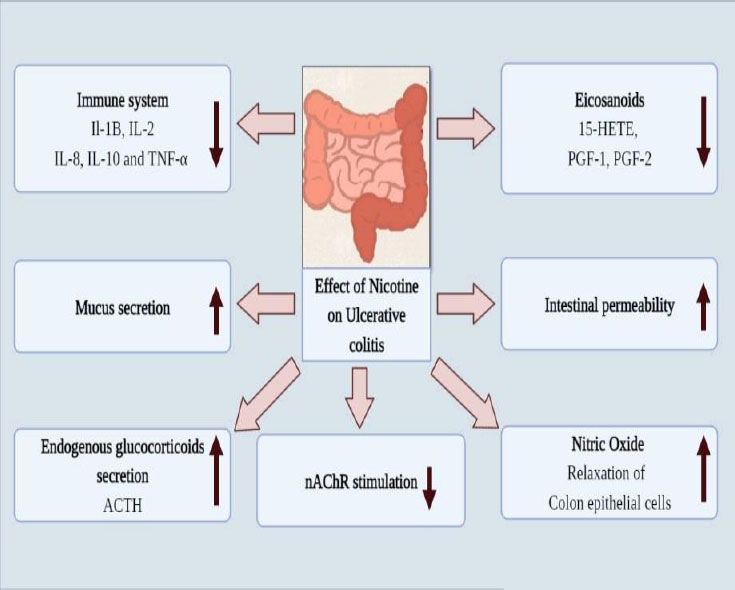
intestinal mucosa [52]. NO is a strong vasodilator that also functions as a nonadrenergic, noncholinergic neurotransmitter, which relaxes the smooth muscles in the sigmoid colon. Endogenous glucocorticoids are hormones that the hypothalamic-pituitary-adrenal axis releases and that are controlled by stress and circadian rhythm. Adrenocorticotropin hormone (ACTH) and other endogenous glucocorticoids, such as glucocorticoids, are more abundant when nicotine is consumed [53]. The elevated endogenous glucocorticoids help ulcerative colitis. Additional possible methods nicotine decreases intestinal permeability and rectal blood circulation, but the significance of these results is uncertain. It has not yet been determined whether nicotine's impact on intestinal permeability and rectal blood circulation is equivalent [54].
2.2.2. Crohn’s Disease
Every region of the GI tract, mouth to antrum, is affected by the chronic inflammatory condition known as Crohn's disease. However, the colon and ileum are most commonly impacted. Crohn's disease exhibits transmural, granulomatous inflammation in the form of an irregular pattern [55]. Three different categories of CD exist- Inflammatory, fistulating, and restricting diseases. Although CD incidence is widespread, some nations, especially those in Western and North America, are more severely impacted than others. Compared to men, women are more frequently affected by CD [56]. According to North American studies, people in the 33.4 to 45 age brackets are the most afflicted by CD. The possibility of CD is greater in Asia and other nations that have been heavily influenced by Western civilization, nutrition, and industrialization. Not enough is known about the CD risk factor. According to different research, the initiation of CD is caused by factors including immunology, the environment, food, and genetics [57]. Nicotine or smoking is an environmental variable that increases the incidence of CD and doubles that risk. In Western nations, it is primarily connected to women. The most significant factor influencing the intestinal gut flora is diet. Diet is the cause of CD, and a drop in nutritional fiber ingestion alters the variety of the gut flora. Appendectomy, low dietary vitamin D, oral contraceptives, postmenopausal hormone, NSAIDs, antibiotics, depression and psychological stress, high dietary fat, and high dietary protein are a few more environmental factors that contribute to CD and immune cells also penetrate the gut of the CD patients [58]. In the process of gut penetration, CD4 T-cells, CD8 T-cells, B-cells, CD14 monocytes, and natural killers are involved—reduction of mucus in the gut by the Muc2 gene causes the CD. FUT2 (fucosyltransferase) is responsible for the secretion of antigen ABO. This FUT2 enzyme decreases the secretion of the antigen, alters the binding with bacteria, and causes the CD. At the location of inflammation, various sticky molecules, including integrin α4β4 and MAcCAM-1, are overexpressed and are crucial in CD [59]. When the number of certain proteins increases in the granular tissue at the site of CD inflammation and revamps extracellular fluid, leucocytes are activated. These proteins include CD44, CD26, and metalloproteins (MMP), being MMP1 and MMP3. The immune system function is also disturbed in CD patients, such as in the overactive T cell, due to this encouraging the Th1 cells by boosting the generation of the cytokines IL-12 and IFN-γ [60]. The generation of TNF-α elevates the numeral of CD4+ FoxP3+ T-REG cells in the mucus membrane of youngsters. According to certain research, Th17 cells coexist with Th1 cells in CD patients and have an impact on the production of IL12, IL23, and TNF-α. Because IL-34 is present at the site of inflammation in CD patients, TNF-α and IL-6 are produced there through an ERK-mediated pathway. Both UC and CD are a part of IBD [61]. In the context of nicotine, it has worsening effects on CD patients while having therapeutic potential on UC patients. Nicotine worsens the signs and symptoms of inflammation in CD patients by triggering the Th1 and Th17 cells, which in turn raises the levels of pro-inflammatory cytokines such as TNF-α, interferon-gamma (IFN-γ), IL-23, IL-6, and IL-1 [54]. Due to the downregulation of colonic CD4 T cell nAChRs controlled by IL-12, nicotine aggravated colonic inflammation with the development of wide-ranging mucosal cell death in the proximal colon, exhaustive crypt injury, entire goblet cell exhaustion, lamina propria ruination, and huge lymphocytic infiltration [62]. In macrophages infected with Mycobacterium avium paratuberculosis, nicotine (4 g/ml) altered TLR2/MyD88/IL-8 and aggravated inflammation, offering crucial information into how nicotine affects the exacerbation of inflammation symptoms in CD sufferers. The impact of nicotine on innate immunity (monocyte/macrophage) in CD is not yet properly known [63].
2.3. Gastrointestinal Cancers
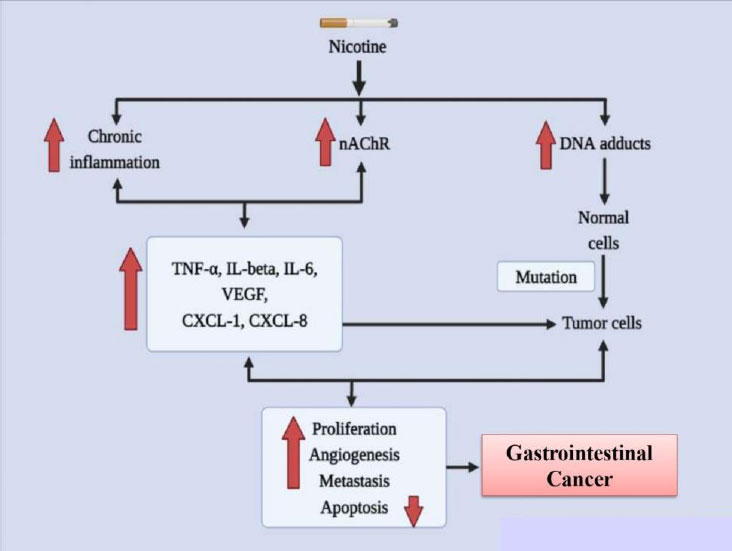
According to the report of the World Health Organization (WHO), nicotine usage killed 100 million individuals in the 20th century and, if it continues unchecked, will kill one billion people in the following century. Additionally, smoking is identified to promote the chances of gastrointestinal (GI) tract malignancies [64]. It boosts carcinogenesis in the gastrointestinal system, liver, and colon and is Taiwan's biggest risk factor for gastric cancer. Strong facts from epidemiological and case-control studies demonstrate that tobacco smoke is an important risk factor for pancreatic cancer, if not the most significant one [65].
Numerous substances found in cigarette smoke have an impact on the GI tract. Since it interacts with nicotinic receptors in the brain, nicotine has been well investigated for its bioactivity on mood, hunger, and goal achievement. It is also highly addictive [66]. Nicotine is swiftly disseminated all across the body after being quickly absorbed through the epidermis, alveoli, and GI tract. The 4-methylnitrosamino-1-3-pyridyl-1-butanone (NNK) and N'nitrosonornicotine (NNN) derivatives of nicotine both exhibit oncogenic properties. Esophageal cancer in rats has been linked to NNN as a cause [67]. In colon adenocarcinoma cells, NNK increases nuclear factor-kappa B (NF- B) affinity as well as the cancer-promoting homomeric nicotinic acetylcholine receptor (nAChR), 7-nAChR. There is growing data that suggests nicotine and its derivatives stimulate and support important mechanisms involved in the development of GI malignancies [68]. Since the 1990s, nicotine has been linked to nearly all GI organ cancers in experimental models employing animals, cell cultures, and clinical data. In this review, we concentrate on nicotine, even though other chemicals present in cigarette smoke also play a role in the growth of cancer [69]. Various factors involved in causing gastrointestinal cancer due to addiction to nicotine are represented in Fig. (3).
2.3.1. Esophageal and Oral Cancer
Epithelial-mesenchymal transition (EMT) of the keratinocyte layer (epithelial tissue) of the esophagus is usually involved with the promotion of mouth and gastroesophageal cancer. Nicotine has been demonstrated to affect the structure of nAChRs in esophageal keratinocyte culturing by dislodging the regional neurotransmitter Ach. Ki-67, PCNA, p21, cyclin D1, and p53 were among the cell cycle and differentiated genes whose transcriptional and translational regulation was altered by nicotine [70]. Nicotine also triggered a shift in the production of the nAChR subunits α3, α5, α7, α2, and α4, as shown by RT-PCR and immunoblotting. The carcinogenic roles of the 7-nAChRs in keratinocyte cultures have been shown in numerous investigations [71]. The NF- κB and SLURP1 expression and efferession, as well as modifications in keratinocyte shape, and subsequent 7-nAChR activation, were shown to be the primary nAChR-receptor-mediated oncogenic processes. Furthermore, nicotine modifies the methylation-dependent regulation of the fragile histidine triad gene (FHIT) to affect gene expression [72]. In cellular assays of squamous cell cancer, promoter methylation of the tumor suppressors p16 and FHIT causes the growth of esophageal squamous cell carcinoma (SCC). Methylation of the FHIT promoter in nicotine-treated cells resulted in modified expression of genes like the anti-apoptotic genes p53 and p16, which promoted growth and proliferative advantages. Additionally, after quitting smoking, esophageal squamous epithelial cells once again expressed the FHIT protein [73].
2.3.2. Gastric Cancer
A significant hazard variable for stomach cancer is nicotine. In vitro research into growth signals and inflammatory messengers has recently revealed molecular details concerning nicotine's stomach cancer-causing abilities. Evidence on the signaling pathways that nicotine uses to accelerate the progression of stomach cancer has been gathered through the innovative experiments carried out in Dr. Cho's laboratory [74]. Through the secretion of COX-2, prostaglandin E2, VEGF, and ERK, as well as the activation of ERK and a COX-2-dependent rise in VEGF and VEGF receptor, nicotine promotes the development and relocation of gastric cancer cells. These discoveries are fascinating because Helicobacter pylori-mediated stomach cancer development uses the same signaling pathways [75]. Investigations into the pathophysiology of gastric cancer cells also focused on the ERK-mediated 5-Lipoxygenase (5-LOX) pathway. In this case, nicotine enhanced the interpretation of 5-LOX to support cell proliferation and invasion. By reducing the proportional transcription of cyclin D1, cyclin E, p-RB, DP-1, and E2F, the protein blocker MK886 that inhibits 5-LOX reduces penetration and causes cell cycle termination [76]. Furthermore, the 5-LOX blockage caused E-cadherin to become inactive and transcriptional repressor Snail to become active, demonstrating an epithelial-mesenchymal transition (EMT). In accordance with this, current research showed that nicotine activation enhanced the number of migrating cells in a transwell migration assay. It has been demonstrated that nicotine causes E cadherin to be markedly downregulated and ZEB-1 and Snail to be upregulated [77]. Additionally, it has been established that nicotine inhibits the interpretation of genes that prevent cell growth by activating a microRNA (miRNA) pathway. One study found that prostaglandin E receptors (EP2 and EP4) and NF-B activation influence the transcription of the microRNAs (miRNA) 16 and 21 in gastric cancer cells [78]. MiRNA-16/21 transcription and cell proliferation were both effectively diminished by NF-B knockdown, while the knockdown of EP2 and EP4 hindered the NF-B/miRNA pathway. Some other investigations discovered a connection between uPAR transcription and clinical manifestations of tumor malignancy in patients with gastric cancer, including tumorigenesis, metastasis, and clinical stage. As nicotine enhances the transcription of uPA/uPAR, it is possible that uPAR could be another clinically important player triggered by nicotine in the development of cancer [79].
2.3.3. Colon Cancer
Nicotine in vitro has indicated important pathways for the development of colon cancer, such as translocation, angiogenesis, multiplication, and escape from apoptosis. By altering the transcription of receptors and their phosphorylation profiles in numerous mitogenic processes, nicotine has been demonstrated to enhance proliferation [80]. Through one investigation, nicotine administration resulted in elevated 5-LOX transcription and phosphorylation of the epidermal growth factor receptor (EGFR) in colon cancer cells. It was possible to prevent the proliferation brought on by nicotine activation by inhibiting these receptors (EGFR and/or 5-LOX) [81]. Similar to this, nicotine also caused the overexpression of acetylcholine and noradrenaline receptors, which boosted colorectal cancer cell multiplication. It has been illustrated that nicotine increases the transcription of 1- and 2-adrenoceptors, which increases VEGF, COX-2, PGE2, and microvessel density. These effects were reversed by atenolol's blockage of -adrenergic receptors [82]. Additional research has shown that nicotine/smoking enhances VEGF, 5-LOX, COX-2, and MMP2/9 in colon cancer, which promotes angiogenesis and neovascularization. By inducing fibronectin, a nicotine7-nAChR signaling pathway has been demonstrated to play a significant role in colon cancer cell migration [83]. In this study, siRNA reduced and pharmacological suppression of COX-2 and 7- nAChR showed that nicotine and COX-2 were required for the induction of fibronectin. Even though nicotine raises the formulation of growth factors and the interpretation of their receptors, autocrine signaling is probably a further significant oncogenic pathway for the onset and evolution of colon cancer. An autocrine pathway for nicotine oncogenesis has been recommended in human colon cancer cells for SLURP-1, though this most likely happens through some of the aforementioned pathways. This brief study found that the indigenous 7-nAChR ligand SLURP-1, which inhibits cell proliferation, is decreased by nicotine [84].
CONCLUSION
Mounting evidence demonstrates that nicotine addiction can induce offensive, defensive, and carcinogenic processes in the GI tract. Nicotine addiction is beneficial in UC, but for other diseases, nicotine addiction is showing harmful effects. Nicotine increases the PGE, mucus, and NO in ulcerative colitis. Gastrointestinal diseases are these factors decrease and cause peptic ulcer, Crohn’s disease, and gastrointestinal cancer. Nicotine mainly affects the nACh receptor. Nicotine increases the caspase cascade mechanism and increases the growth of GI diseases. Normal cells are converted into tumor cells due to mutation in the DNA adducts and cause GI cancers.
LIST OF ABBREVIATIONS
| COPD | = Chronic obstructive pulmonary disease |
| GI | = Gastrointestinal |
| NSAIDs | = Non-steroidal anti-inflammatory drugs |
| H. pylori | = Helicobacter pylori |
| nAChRs | = Neuronal nicotinic acetylcholine receptors |
| cAMP | = Cyclic Adenosine Monophosphate |
| MUC | = Mucin |
| MUC5AC | = Mucin 5AC |
| EGF | = Endothelial growth factor |
| NO | = Nitric Oxide |
| VEGF | = Vascular endothelial growth factor |
| cNOS | = Constitutive nitric oxide synthase |
| nNOS | = Neuronal nitric oxide synthase |
| iNOS | = Inducible nitric oxide synthase |
| eNOS | = Endothelial nitric oxide synthase |
| PGE 2 | = ProstaglandinE 2 |
| IBD | = Inflammatory bowel disease |
| UC | = Ulcerative Colitis |
| CD | = Crohn’s Disease |
| CD4+ | = Clusters of differentiation 4 |
| Th | = T helper cells |
| INFγ | = Interferon gamma |
| IL | = Interleukin |
| NKT | = Natural killer cells |
| TNF | = Tumor necrosis factor |
| 15 HETE | = 15-hydroxy eicosatetraenoic acid |
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
There is no conflict of interest.
ACKNOWLEDGEMENTS
We are thankful to the Director of Pranveer Singh Institute of Technology (Pharmacy), Dr. A.K. Rai, for the support and motivation.

